The EPPVDN – Our Agenda
Learn more about our international investigators and the latest news on PH and heart failure…
Learn more about our international investigators and the latest news on PH and heart failure…
Learn more about our international investigators and the latest news on PH and heart failure…
Follow us on Twitter!
Challenges and Future Directions of the European Pediatric PVD Network
Challenges and Future Directions of the European Pediatric PVD Network
Follow us on Twitter!
PH and Heart Failure – we are searching for a Cure
The European Pediatric Pulmonary Vascular Disease Network is a non-profit organization that strives to define and develop diagnostic methods and new therapies (pharmacotherapy, critical care, VAD/ECMO, cahter/surgical interventions, bilateral lung transplantation) in all forms of pediatric hypertensive vascular disease (PHVD):
The forms of PHVD include pulmonary arterial hypertension (e.g., idiopathic PAH, hereditary PAH, and pulmonary hypertension (PH) associated with congenital heart disease (PH-CHD)), PH associated with chronic lung disease (PH-nCLD/bronchopulmonary dysplasia), persistent pulmonary hypertension of the newborn (PPHN), pulmonary venoocclusive disease (PVOD), pulmonary capillary hemangiomatosis, among others. Moreover, the network adresses the detrimental cardiac dysfunction and heart failure associated with PH, in particular, the RV – pulmonary artery (PA) unit, right ventricular failure, RV – LV interaction and LV compression, right (RA) and left atrial (LA) hypertension.

About our Work
To achieve the above, the European Pediatric Pulmonary Vascular Disease (PVD) Network members meet on a regular basis, develop joint preclinical and clinical studies, patient registries, and the first multipaper expert consensus statement on the diagnosis and treatment of pediatric pulmonary hypertension. If you like to support our work, please go to „Get involved“ or contact us via email.

About PVD and RV Failure
Pulmonary vascular disease (PVD) is characterized by progressive obliteration of pulmonary arterioles leading to increased pulmonary vascular resistance (PVR), pulmonary arterial hypertension (PAH), right heart failure, and death in ≈25-60% of patients 5 years after diagnosis. The pathobiology of PAH is complex and multifactorial, and none of the current therapies has been shown to be universally effective or able to reverse advanced pulmonary vascular disease, characterized by plexiform vascular lesions.
Right Ventricular Failure – the #1 Cause of Death in PAH

CO, cardiac output; PAP, pulmonary artery pressure; PVR, pulmonary vascular resistance
Untreated idiopathic PAH (IPAH) results in death within 1-3 years after diagnosis due to RV failure and low cardiac output. The 5-year survival for children with IPAH is only 75%, with a freedom from death or transplantation of only 57%. Lung or heart-lung transplantation is far from ideal, with a 5-year post-transplant survival of only ≈ 45%. Conversely, PH is a common complication of epidemic left heart diseases (LHD), and RV dysfunction (RVD) is common in (left) heart failure with preserved ejection fraction (HFpEF in LHD). The “dogma” that (a) RV dysfunction is exclusively a direct consequence of PA pressure elevation in PAH, and (b) targeted therapy does not need to directly address RV dysfunction in either PAH or HFpEF in left heart diseases, has recently been challenged.
Increasing number of Publications on PAH therapies from 1990 to 2019
In the last 30 years, rapid, accelerated knowledge gain due to an increasing number of publications on pathobiology and clinical trials have led to more tailored (“personalized”), goal oriented treatment modalities for PAH. New insights into metabolic dysregulation and previously unknown gene mutations, and high impact, high volume, longterm randomized controlled trials (RCT) using combined outcomes such as “first morbidity and mortality”, have been published, leading to the approval of more PAH drugs acting on previously known pathways. However, prospective studies in children and adolescents with PAH are largely lacking.
The number of publications addressing experimental therapies in PAH began to increase in 1995 after landmark studies in human PAH tissues and randomized controlled clinical PAH trials had been published.
Legend:
To evaluate the number of publications addressing experimental therapies in pulmonary arterial hypertension (PAH), the search engine Web of Science, which is a citation database of research literature, was used (www.webofknowledge.com). In the advanced search options we refined our outline query to Pulmonary Arterial Hypertension AND therapies and analyzed the Number of publications within the years 1990 and 2019.
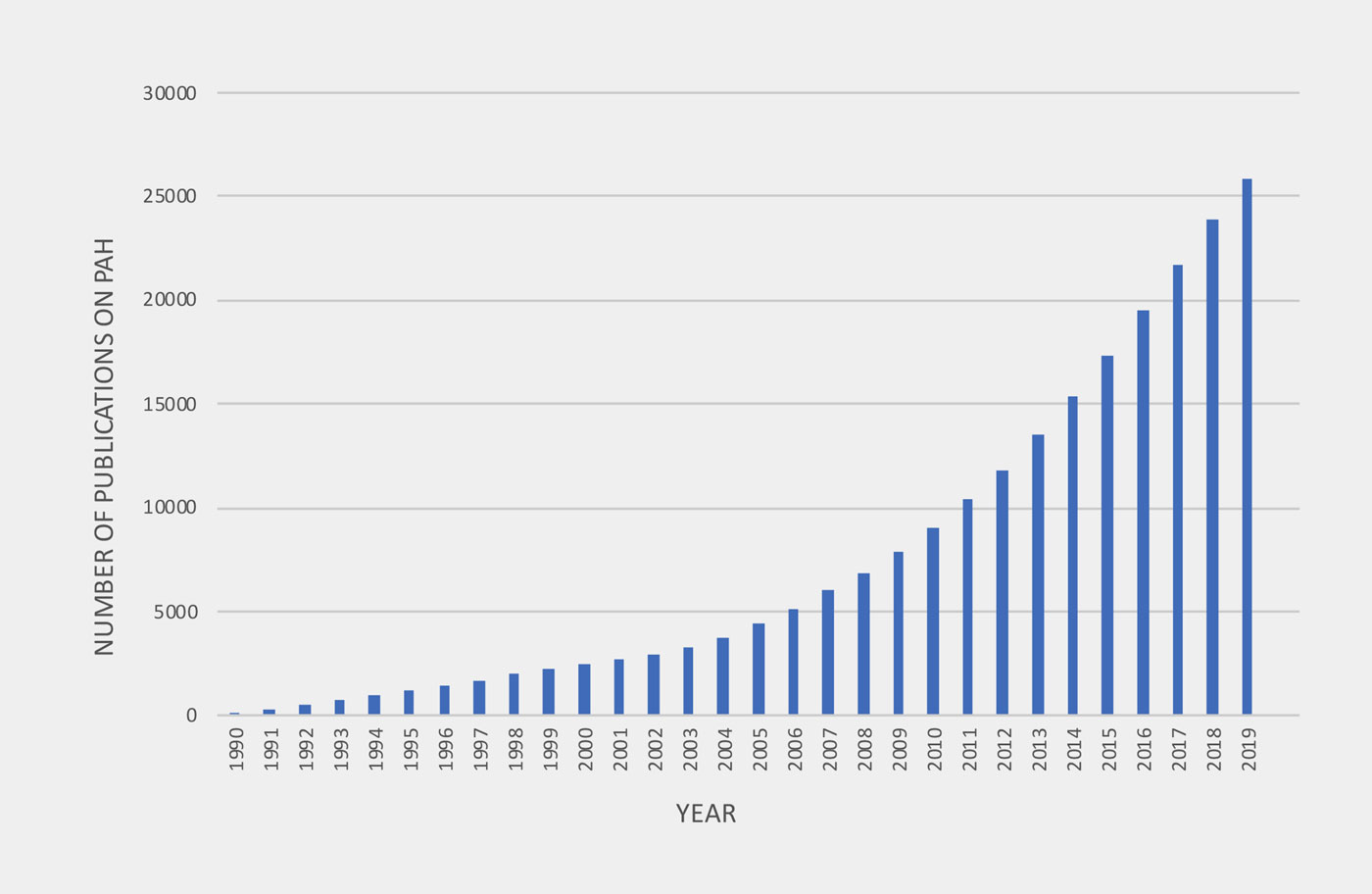

In the last 30 years, rapid, accelerated knowledge gain due to an increasing number of publications on pathobiology and clinical trials have led to more tailored (“personalized”), goal oriented treatment modalities for PAH. New insights into metabolic dysregulation and previously unknown gene mutations, and high impact, high volume, longterm randomized controlled trials (RCT) using combined outcomes such as “first morbidity and mortality”, have been published, leading to the approval of more PAH drugs acting on previously known pathways. However, prospective studies in children and adolescents with PAH are largely lacking.
The number of publications addressing experimental therapies in PAH began to increase in 1995 after landmark studies in human PAH tissues and randomized controlled clinical PAH trials had been published.
Legend:
To evaluate the number of publications addressing experimental therapies in pulmonary arterial hypertension (PAH), the search engine Web of Science, which is a citation database of research literature, was used (www.webofknowledge.com). In the advanced search options we refined our outline query to Pulmonary Arterial Hypertension AND therapies and analyzed the Number of publications within the years 1990 and 2019.
Molecular Targets of Future Pulmonary Hypertension Therapies Adressing RV Dysfunction (2020)
By July 2020, more than 10 drugs have been approved for the treatment of adult PAH, all of which are primarily vasodilators. More than 35 randomized controlled trials (RCT) have shown a moderate effect of these agents (PDE5 inhibitors, ERA, prostacyclin analogues) – based on short-term improvement in functional capacity or longer time to clinical worsening. Hemodynamically, the drugs tend to increase cardiac output but have only little effect on elevated PA pressure and RV dysfunction. Although recent PAH studies on early combination therapy and new targets in classical signaling pathways (guanylate cyclase, IP receptor) have raised great hope for better composite outcomes, until today, no RCT lasting longer than 16 weeks has shown a decrease in mortality as single primary outcome.
Legend:
Summary of metabolic derangements identified in RV cardiomyocytes in PAH. Solid arrows denote pathways increased in PAH, and dashed arrows represent pathways decreased in PAH. RV, right ventricular; PAH, pulmonary arterial hypertension.
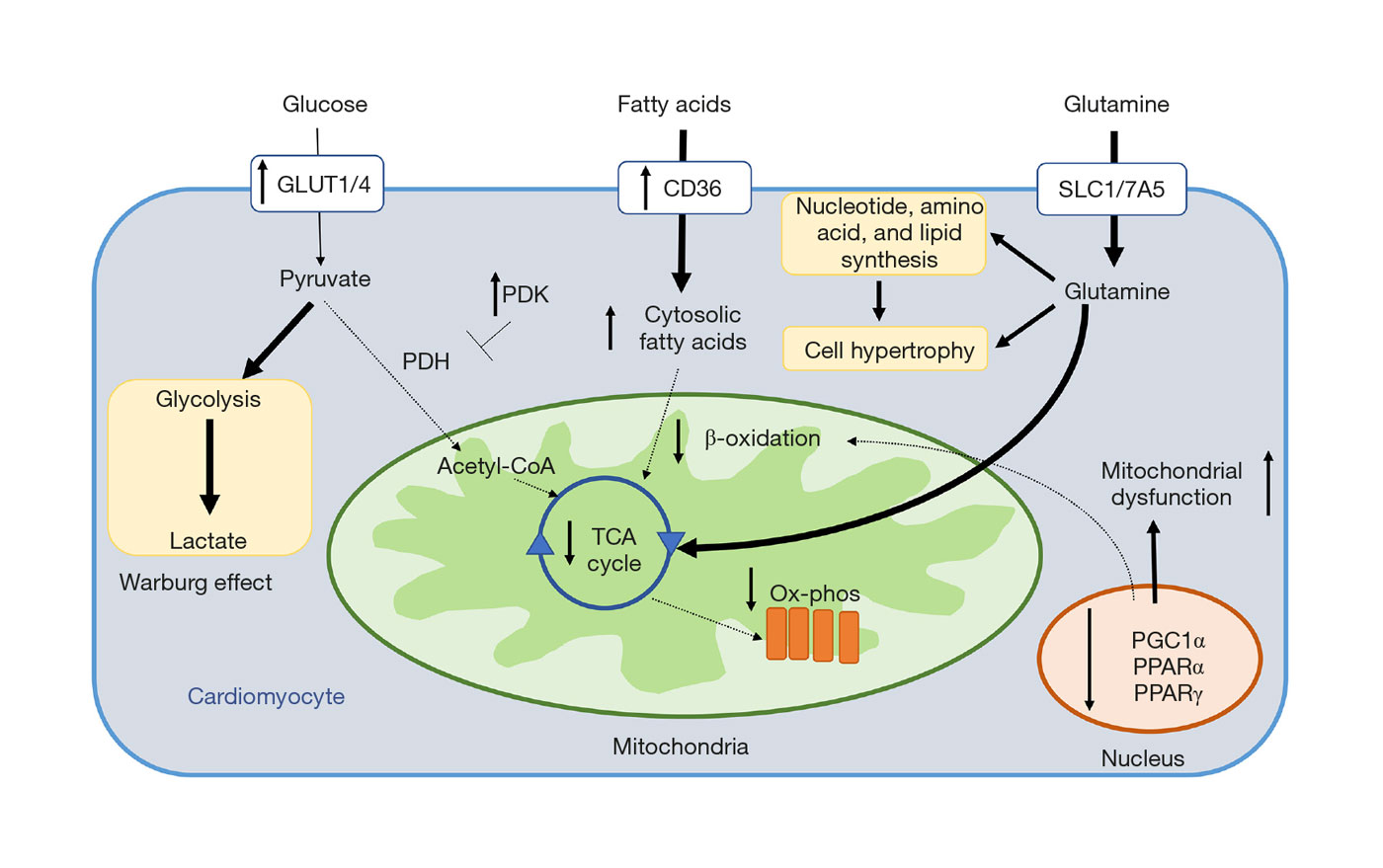
From: Vineet Agrawal, Tim Lahm, Georg Hansmann, Anna R. Hemnes. Molecular mechanisms of right ventricular dysfunction in pulmonary arterial hypertension: focus on the coronary vasculature, sex hormones, and glucose/lipid metabolism. Cardiovasc Diagn Ther 2020.
Ongoing Clinical PAH Trials in 2013
Administration of locally acting (inhaled) or chemically modified, previously tested agents could likely circumvent or limit the problem of systemic adverse effects. Drug development should generate targets for HFpEF/PH-LHD and PH-RVD, and systemically given agents should address RV dysfunction and RV-LV interaction directly. Translational research is thus essential to develop new biomarkers and reverse-remodeling strategies for the treatment of pulmonary vascular disease, pulmonary hypertension and heart failure through preclinical mechanistic in vivo studies using animal models resembling human PAH, and subsequent early tailored clinical pilot studies.

This leads to 21 Industry-funded trials and 44 Non-industry-funded-trials.
Industry-funded Trials

© Gopinath Sutendra and Evangelos D. Michelakis Sci Transl Med 2013;5:208sr5
Non-industry-funded Trials

© Gopinath Sutendra and Evangelos D. Michelakis Sci Transl Med 2013;5:208sr5
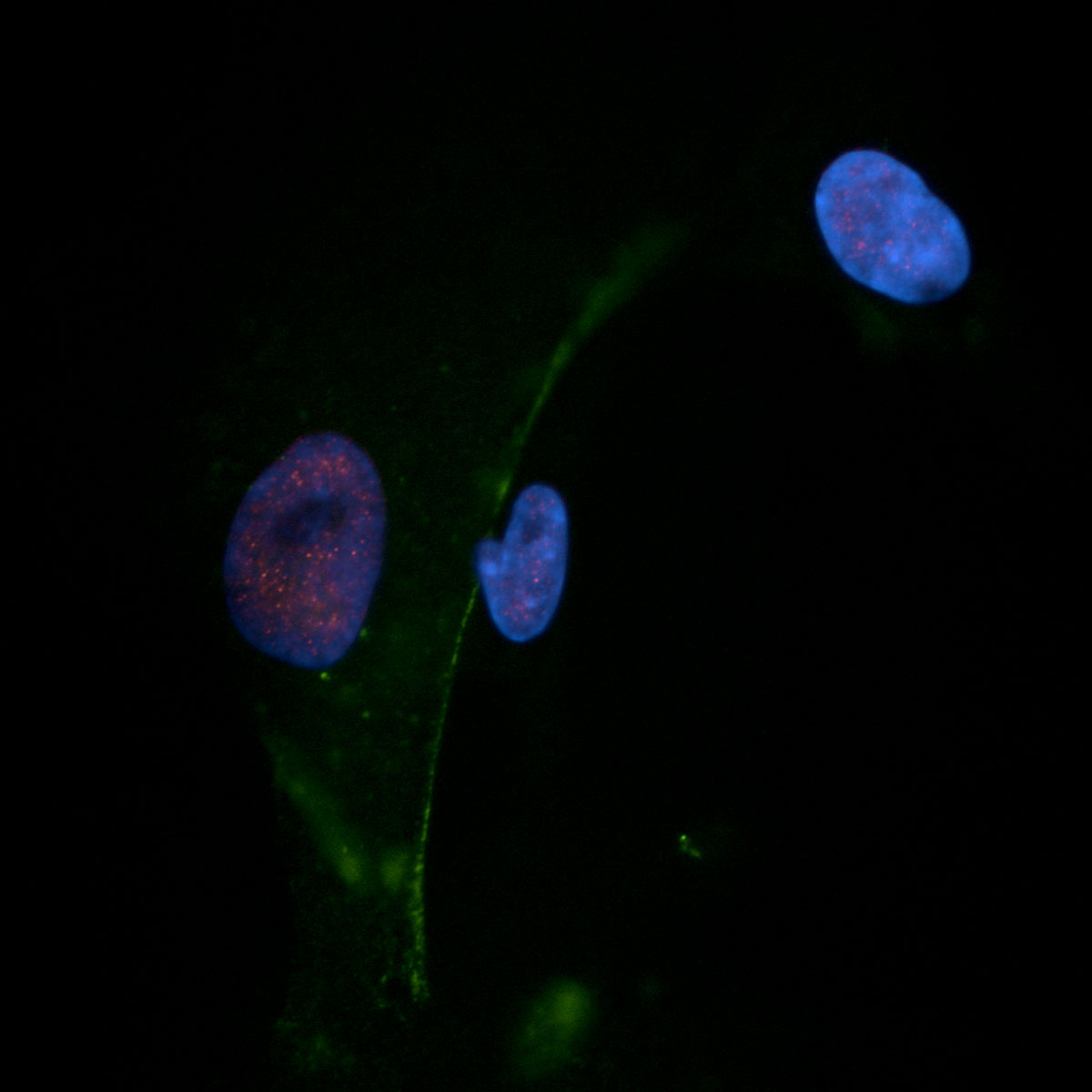
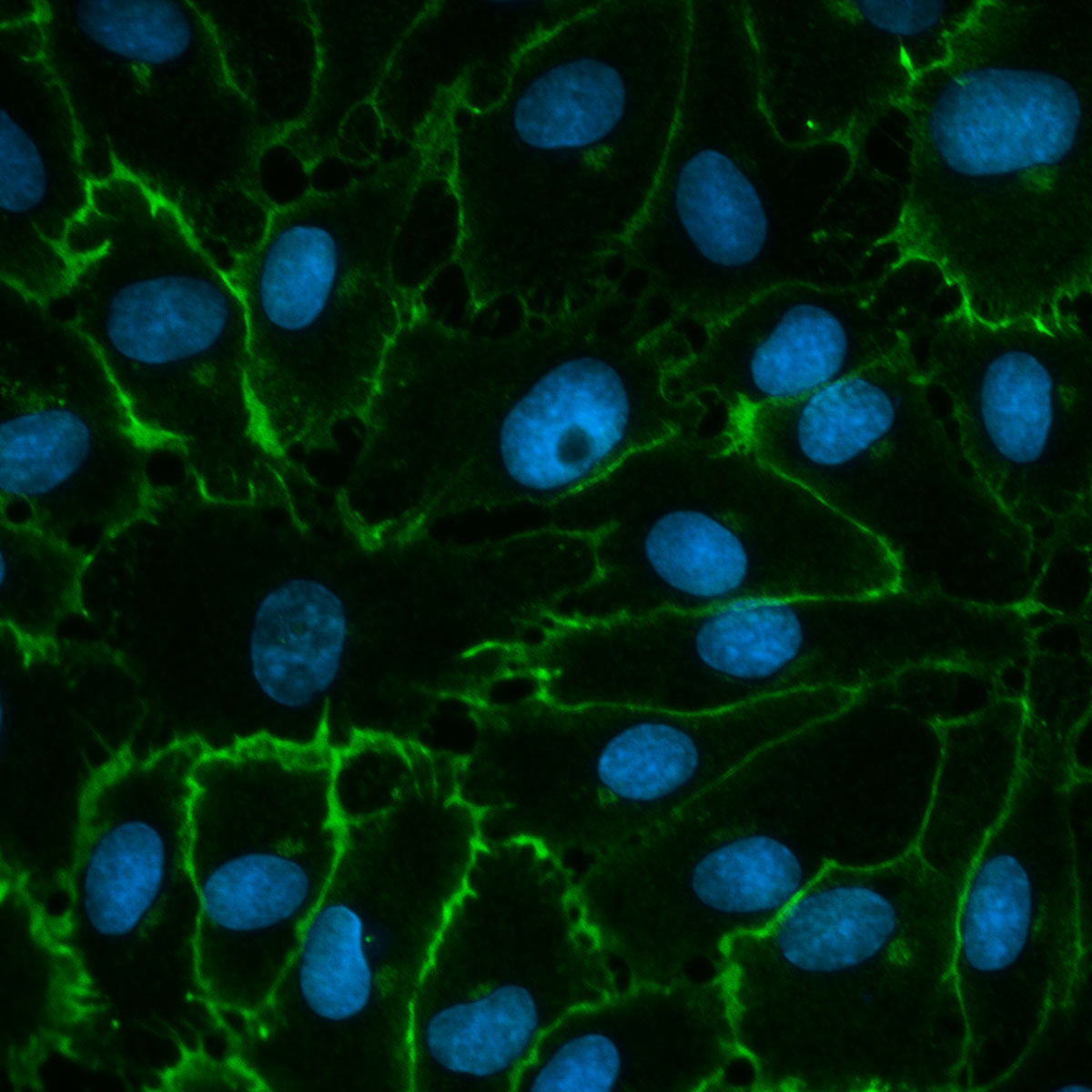
Cardiovascular Cells
First Image:
Human pulmonary artery smooth muscle cell (Lamin A/C, DAPI)
Second Image:
Explanted, isolated and subcultured human pulmonary artery endothelial cells from a patient with endstage idiopathic PAH (VE-cadherin, DAPI)